

#ScienceSaturday posts share exciting scientific developments and educational resources with the KAND community. Each week, Dr. Dylan Verden of KIF1A.ORG summarizes newly published KIF1A-related research and highlights progress in rare disease research and therapeutic development.
KIF1A-Related Research
KIF1A neurodegenerative disease mutations modulate motor motility and force generation
One challenge in researching KAND is that a patient’s symptoms can vary so much depending on the type and location of the KIF1A mutation; for every study investigating the impacts of a KIF1A mutation, we have to ask how much we can generalize the findings to other mutations, or the people affected by them.
This is why direct comparisons of different KIF1A mutations are invaluable; our patient-derived iPSC cells were developed for such comparisons, and projects like the Natural History and EEG studies help us find clinical patterns in KAND, so we can target therapies to the appropriate patients. But with over 100 identified KIF1A mutations, we still have a lot to learn about the similarities and differences between them.
In this week’s pre-print*, researchers at the Indian Institute of Technology Gandhinagar and Sambalpur University in India compared the impact of 16 KIF1A mutations in a battery of experiments to look for patterns of KIF1A dysfunction.
Testing KIF1A mutants in multiple cell types
This study introduced KIF1A mutations to two types of cells:
- COS-7 cells are non-neuronal, but have a rich microtubule network that extends from the cell’s center to its edge, allowing for easy assessment of KIF1A movement.
- CAD cells can differentiate into neuron-like cells, with long processes similar to axons. This can serve as a better model for neuronal KIF1A, including effects of KIF1A on neuronal development.
Using both cell lines has two benefits: First, they can provide slightly different readouts of KIF1A activity. Second, when we see similar KIF1A behavior in two separate cell types, that information is more reliable.
The researchers assessed KIF1A’s speed, distribution, and ability to transport both small and large cargo across these 16 mutations.
KIF1A localization and transport
Healthy KIF1A typically moves from the center of the cell outward; in COS-9 cells this means moving toward the edge of the cell, and in CAD cells this means toward the end of the axon-like tip, called a neurite. In these tests, KIF1A mutations fell into one of three categories
- Peripheral mutations (V144F, V220I, E233D, and A255): These mutations didn’t prevent KIF1A alone from reaching the edge of the cells, but did have mild impact on cargo transport. These variants were of uncertain significance or had milder clinical symptoms.
- Cytoplasmic mutations (S58L, A202P, R216P, R216H, L249Q, T312M, R316W, and R350G) caused KIF1A to move more slowly and fall off of the microtubule more often, causing moderate issues with transport of small and large cargo. This group included mutations that have a wide variety of clinical manifestations.
- Microtubule binding mutations (T46M, T99M, G102D, S215R, and E253K) caused KIF1A to stick to microtubules with very little movement, completely preventing the transport of cargo and potentially causing roadblocks on the microtubules. Notably, several of these mutations prevented CAD cells from developing axon-like structures, indicating an impact on neuronal development. Some of these mutations like T99M and E253K are known to be quite severe, while others like G102D are associated with milder symptoms.
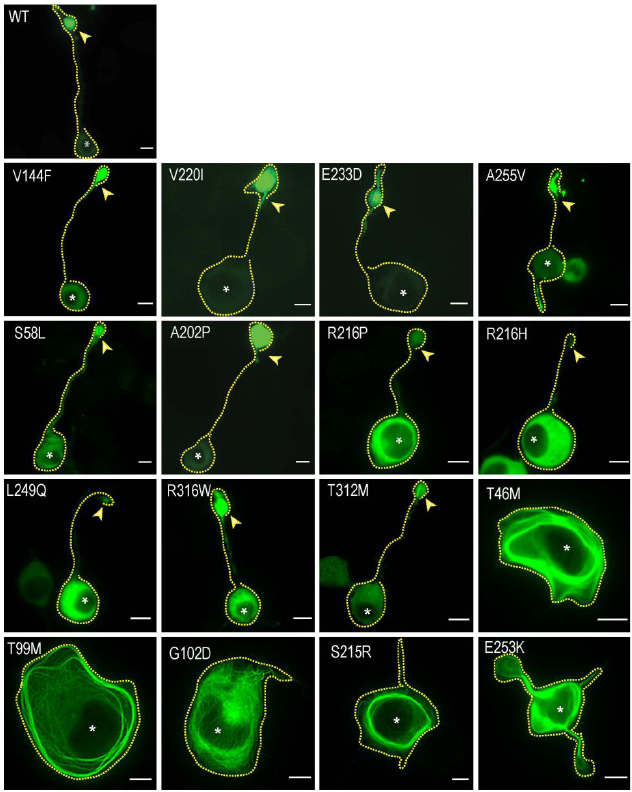
As you can see in the figure above, there is variability even between mutations in the same category, and the correlation between these categories and disease severity isn’t absolute; relating these data back to clinical findings is crucial. But with these findings and knowledge of KIF1A structure, researchers can identify trends and commonalities among different mutations, and hopefully consolidate therapeutic approaches for KIF1A mutations with similar traits.
*What’s a pre-print? Check out this #ScienceSaturday post to learn more.
Rare Roundup
NeuCyte presents KAND model work at the International Society for Stem Cell Research conference
This week, our partners at NeuCyte presented their work in developing cellular screening platforms for KAND, based on patient-derived stem cells. This work included the identification of neurodevelopmental and axon growth deficits, as well as seizure-like activity; these readouts can be used to assess potential therapeutics for KAND and accelerate our therapeutic pipeline. Want to learn more about NeuCyte’s work? They will be presenting at our 2023 Family and Scientific Engagement Conference!

Research to develop new rare disease therapies underway at The Jackson Laboratory
In the rare disease space, it takes a village – only through collaboration between clinicians, researchers, regulators, and patients can we develop effective treatments for devastating disorders. It was through such collaboration that KIF1A.ORG’s 2017 “We need a mouse” campaign resulted in the beginning of a KIF1A program at the Jackson Laboratories (JAX), who went on to create mouse lines carrying KIF1A mutations for researchers to study KAND.
The FDA and NIH have taken several recent steps to better serve rare and genetic disorders, including the creation of Operation Warp Speed and attempts to create standardized gene therapies. This week, the NIH has also provided funding to Cat Lutz’ team at JAX as part of the Somatic Cell Gene Editing Program; the team aims to de-risk and develop gene editing therapies by testing them preclinical mouse models. The initial targets for this project are Huntington’s Disease, Friedrich’s Ataxia, Rett Syndrome, and Spinal Muscular Atrophy – these will serve as proofs of concept for JAX’s approach to treat genetic neurodevelopmental and neurodegenerative disorders.