

#ScienceSaturday posts share exciting scientific developments and educational resources with the KAND community. Each week, Dr. Dylan Verden of KIF1A.ORG summarizes newly published KIF1A-related research and highlights progress in rare disease research and therapeutic development.
KIF1A-Related Research
Genetic landscape of congenital insensitivity to pain and 2 hereditary sensory and autonomic neuropathies
KAND heterogeneity – the diversity of symptoms experienced by KAND patients – is a constant consideration. Providing treatments for our community means understanding the different ways KAND can manifest, and learning from other disease groups with overlapping symptoms.
One subtype of KAND is called HSAN (hereditary sensory and autonomic neuropathy) KAND; this is caused by loss-of-function mutations affecting the cargo-binding domain of KIF1A, and patients experience late-onset loss of sensation, including pain. Pain isn’t a pleasant sensation, but it plays a huge role in our survival: without pain, we may not tend to injuries or remove noxious dangers from our environment.
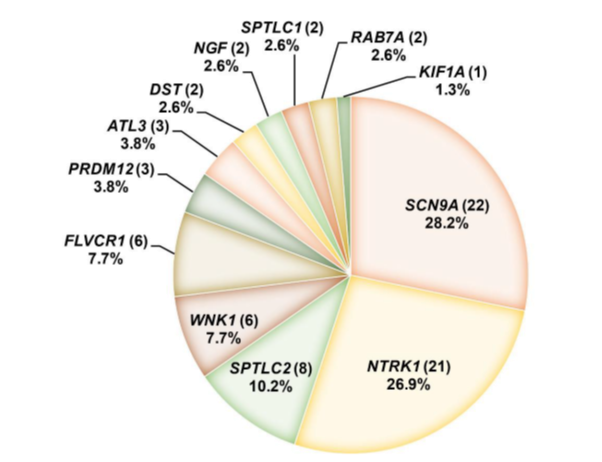
In this week’s article, a multinational team of scientists investigated the genetics of patients with HSAN or a similar disorder, congenital insensitivity to pain. The study looked at 78 individuals experiencing reduced sensitivity to pain.
The study only identified one individual with a KIF1A mutation, which was located in the cargo-binding domain of KIF1A. While this is a small piece of the pie, we can look at how other genes related to HSAN/pain insensitivity interact with KIF1A function. For example:
- NGF and NTRK1: NGF is a growth factor that activates the receptor NTRK1, a KIF1A cargo. These molecules regulate neuronal growth and survival, and mice with insufficient KIF1A show NTRK1 transport defect and reduced pain sensitivity.
- WNK1: WNK1 is a kinase that interacts with KIF1A and may regulate its ability to unload cargo at axon tips.
- SCN9A/Nav1.7: Nav1.7 is a sodium channel that allows neurons to fire, and is enriched in pain-sensing neurons. While it hasn’t been directly paired to KIF1A, it is transported to axon tips along microtubules.
Studies like these that reassess umbrella disorders for genetic contributions are a two-fold advantage; we can use overlap between disease subtypes to search for common therapeutic approaches, and use distinctions between subtypes to suss out the leads most likely to apply to our smaller community.
Rare Roundup
Gene therapies for rare diseases are under threat. Scientists hope to save them
When working on complex and multi-stage projects, the reward for solving one problem is often a new set of challenges. As we advance our mission to find treatments and cures for KAND, we are also turning our minds toward questions of accessibility: A treatment is no good if it can’t get to the patient who needs it.
For rare disease groups, this challenge can become a funding bottleneck, wherein the drug has been made but can’t be produced profitably by a company. This was the case for Strimvelis, a gene therapy for a rare immune disorder; the treatment was highly effective, but still abandoned by two companies who couldn’t sustain its production and administration. This year, the Italian non-profit the Telethon Foundation announced that it would begin producing Strimvelis. This proof-of-principle attempt is one of many possible solutions to the economic challenges of industrializing rare disease treatments and cures. Thanks to broader rare disease organizers like CZI and GlobalGenes, rare disease groups like ours have platforms to compare approaches, results, and create best practices to get novel treatments to people in need.