

#ScienceSaturday posts share exciting scientific developments and educational resources with the KAND community. Each week, Dr. Dominique Lessard and Dr. Dylan Verden of KIF1A.ORG summarize newly published KIF1A-related research. From February 5 – April 16, 2022, a team of talented students from Columbia University’s M.A. in Biotechnology program is taking over the Rare Roundup section of the #ScienceSaturday blog! What topics do you want to learn more about? Send suggestions to our team at impact@kif1a.org.
KIF1A-Related Research
Finding a cure for the rare but severe KIF1A Associated Neurological Disorder, what we (need to) know
Looking for a great summary discussing KIF1A biology, KAND basics, as well as potential KAND treatments and cures? Look no further! This week we are featuring a review-style paper out of the Utrecht University in the Netherlands that covers all the above topics and more. We think this is a great resource to help us understand the big picture history of our KIF1A understanding and how the discovery of KIF1A mutations have shaped and influenced the field of KIF1A research over time.
Diving in further, this paper breaks down what we know about a select handful of mutations—from how these mutations impact KIF1A function on a molecular level to how they may present on a clinical level—and how this information can inform us about potential treatments for KAND. There are many treatment questions and hypotheses discussed in this article such as (but not limited to): Can other kinesin proteins help relieve deficits in KIF1A cargo transport? Can we find small molecule drugs that can bypass the mutated section of the KIF1A protein? Click the button below to read this paper and learn more. Thank you to author Phebe Vermeer for your thought-provoking synthesis of the KIF1A and KAND literature! Want to brush up on small molecule drugs and how they could be utilized to treat KAND? Enjoy the video below!
Rare Roundup
Welcome to the #ScienceSaturday Takeover portion of today’s post! Meet our guest bloggers from Columbia University, Aaron, Pragya, Keyue, Rakshitha, and Hazel, here.
The effect of the pandemic on patients with intellectual and developmental disorders
With the COVID-19 pandemic affecting various aspects of our lives, it is crucial to be aware of the data available regarding the rate of hospitalizations in patients with intellectual and developmental disabilities (IDD). Many diseases differentially affect individuals with preexisting conditions, including COVID-19. The article highlights the severity of this situation in terms of intensive care unit (ICU) admission, invasive mechanical ventilation, 30-day readmission, all-cause in-hospital mortality, and increased length of stay using large electronic administrative databases with information from 900 hospitals in the United States. The study uses statistical data analysis tools to compare models of IDD patients and patients without disabilities. The results signify that COVID patients with IDD were generally younger male patients and those with down syndrome were more likely to face the most severe repercussions, while autistic patients saw lengthier hospitalization periods out of all the patients with IDD. The authors conclude: “Results suggest that patients hospitalized with COVID-19 with IDD have a significantly increased risk of severe outcomes, 30-day readmission, and longer length of stay.” This study’s findings add evidence to consider individuals with IDDs as a high-risk population to prioritize for vaccines and emerging therapies.
Always discuss any questions you have about COVID-19 and vaccinations with your doctor.
Record-breaking rapid DNA sequencing promises timely diagnosis for thousands of rare disease cases
Rare disease families know it usually takes years to receive a correct diagnosis.
With the recent progress in genome sequencing and computational innovation, the price and the turnaround time for genetic testing has reduced drastically. The Human Genome Project, the first successful attempt to sequence a complete human genome, took 13 years and cost $2.7 billion. In 2014, a full genome sequence cost $1,000!
In their quest for a Guinness world record, Stanford researchers used a DNA sequencing platform that reads genomes by pulling large strands of DNA through pores, generating a subtle electric signal unique to each DNA letter. This allowed them to read the entire genome in a record time of 5 hours, 2 minutes. The ultra-rapid DNA sequencing generated terabytes of data that utilized a cloud-based storage system and algorithms to scan the genome for tiny variations (mutations) within the DNA. As this technology becomes more accessible, more rare disease patients will benefit from quicker, more accurate diagnoses.
Scientists Create Vast Data Resource to Uncover ALS Subtypes
Led by researchers at Cedars-Sinai, more than 100 scientists have co-established a new data resource and web-based tool called Answer ALS. The team collected blood samples from over 1,000 ALS patients to create induced pluripotent stem cells (iPSCs) to model the disease. Information about these iPSCs is available to the scientific community in the web-based tool. Now, researchers can uncover new subtypes of ALS. The scientists point out that this resource can help find new treatments for ALS and lead to the development of new drugs that target specific cells and pathways in certain subgroups.
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease, which causes damage in nerve cells of the brain and spinal cord. In this effort, scientists used the iPSCs to create neuron cells of the spinal cord. On the basis of these cell models scientists are using molecular techniques to look for and analyze the proteins that might be affected by ALS, in a massive scale that has not ever been seen before.
“This was a huge collaborative effort, and we hope the information we collected will help lead to the discovery of new molecular subtypes of ALS. With this knowledge we may at last be able to develop drugs targeted to these subtypes, laying the groundwork for new and improved therapies.”
Clive Svendsen, PhD, Co-Director of Answer ALS
With CRISPR gene editing, unique treatments begin to take off for rare diseases
With CRISPR at the forefront of novel rare disease therapeutics, this article dives into some of the individual stories, promise and limitations of the new technology. The article discusses the case of Paddy Doherty who, through the passing of his father, discovered he suffered from transthyretin (ATTR) amyloidosis, a genetic rare disease that causes overproduction of a misfolded protein in the heart. Though the advent of CRISPR/Cas9 came too late for his father, a CRISPR therapy has him back to normal health, undertaking hikes that his condition had previously left him unable to do.
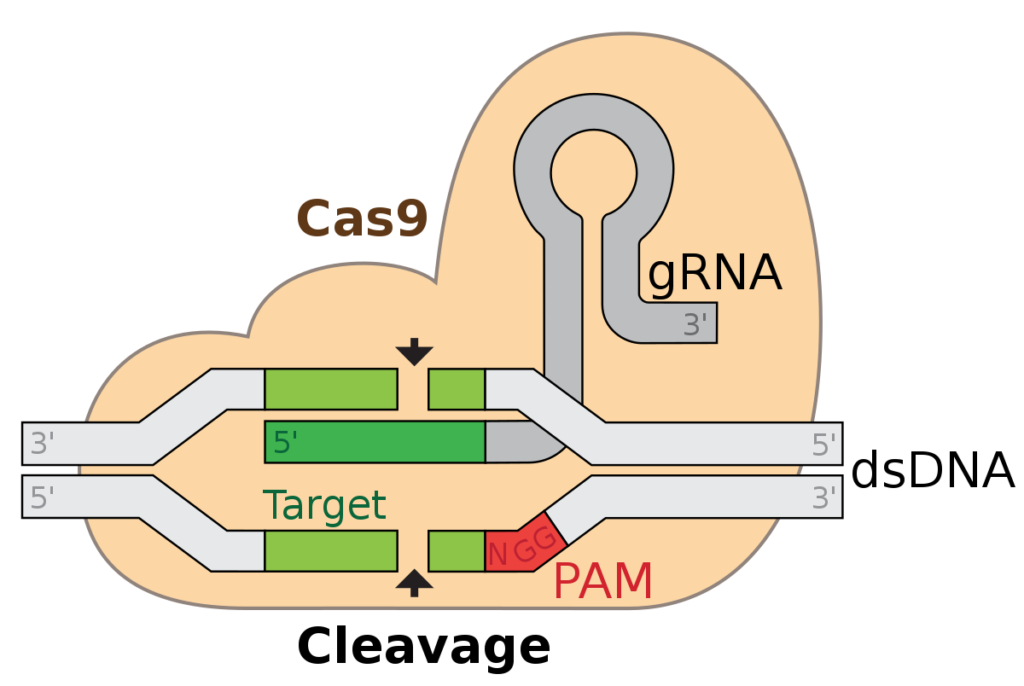
Before we continue… what is CRISPR?
CRISPR/Cas9 acts like a pair of genetic scissors that cuts the cell’s DNA at a specified point, disrupting the gene. If provided with a DNA template, CRISPR/Cas9 can repair a cell’s DNA and incorporate a new change. This allows the replacement of variants that cause disease with a new template, or as with ATTR amyloidosis, disruption of the damaging gene. In this way, the technology holds promise for many genetic rare diseases, because it allows replacement or deactivation of the defective copy of a gene.
How does CRISPR work? The guide RNA (gRNA) tells the Cas9 protein where to cut (cleave) in the cell’s DNA (labeled here as double-stranded DNA, “dsDNA”). Once the DNA is cut, the cell will try to repair it. This can be done with a DNA template (different from the gRNA and dsDNA in the illustration), which can include a desired change. Or, it can be done without a template, which will usually include mistakes, “deactivating” the gene.
Now back to the article…
Doherty was treated by the infusion of CRISPR molecules directly into his bloodstream. Here, these CRISPR molecules could disrupt the gene that was responsible for the overproduction of misfolded protein that is characteristic of ATTR amyloidosis. What makes this trial incredibly promising is that direct infusion of CRISPR molecules into the bloodstream was safe and effective for Doherty. Though an important step in the development of CRISPR therapeutics, this technique won’t be applicable for every disease. Different diseases will require different modes of administration to specifically treat cell types affected by the disease.
For example, ongoing clinical trials targeting sickle cell disease entail the removal of patient stem blood cells. These are then treated with CRISPR to change the DNA, and re-infused into the patient. Because KAND is a neurological condition that affects the nervous system, treatment may require a different delivery system. A crucial step in developing CRISPR therapeutics is making sure the CRISPR molecules get access to the right cell types. Direct blood infusions may not work because the bloodstream is separated from the brain by the blood-brain barrier, which is often difficult to cross for drugs, and especially for proteins (which are much bigger), like Cas9. Equally, to avoid off-target effects (where CRISPR cuts the wrong stretch of DNA), CRISPR molecules should get as little access as possible to the wrong cell types. Different diseases affecting different cell types will thus require different delivery systems, all of which must be developed.
Ultimately, CRISPR/Cas9 is an incredibly exciting tool with the example described representing an inspiring story in the rare disease community, and an important step in the development of CRISPR therapeutics. Nonetheless, CRISPR needs more advancement before it can address the wide variety of rare diseases that are present (this topic is addressed in the 18th of December #ScienceSaturday, if you are interested!).
CRISPR-Cas9 can give rise to undesired, inheritable mutations across the genome
It sounds fascinating that we could tailor CRISPR to directly correct mutations in the KIF1A gene and even cure KAND if the functions of neurons can go back to normal as well. The last article highlighted this potential, and you may be wondering now why such a wonderful treatment is not yet available for KAND patients. This is because CRISPR is not perfect in the whole picture. It may sound disappointing, but the good news is that emerging technologies can help us identify challenges and then try to fix them. The latest research summarized below is a good example.
Traditionally, we sequence DNA in short fragments and piece them together, like recovering a torn-up book chapter, to obtain the whole sequence. During this process, some pieces are left out, and errors are more likely to happen if the “book” is big—imagine your genome is 3 billion letters long and you can only read 300 at a time. A new sequencing technology called “Nanopore” is able to read long stretches (about 30,000) of DNA and generate more complete and reliable results, which is used by the authors of this paper to evaluate the effects of CRISPR-Cas9 on zebrafish. In addition to mutations introduced on the target genes, undesired mutations of various types were also detected in the edited genomes, especially large deletions and insertions that were not reported by previous studies. These mutations are termed “off-target” because they can occur in genes with normal functions rather than those responsible for the disease, causing new abnormalities in the cell. We already know very well how unwanted mutations in the normally functional KIF1A gene give rise to KAND—fixing mutations in KIF1A while introducing mutations in other genes could present a real risk. This is something we need to be cautious about before applying CRISPR-Cas9 to human bodies. Improving sequencing methods like “Nanopore” could help us better validate the efficacy and safety of CRISPR-based gene therapy in both preclinical settings on animal models and clinical trials on humans.