

#ScienceSaturday posts share exciting scientific developments and educational resources with the KAND community. Each week, Dr. Dylan Verden of KIF1A.ORG summarizes newly published KIF1A-related research and highlights progress in rare disease research and therapeutic development.
KIF1A-Related Research
Genetic and clinical features of pediatric-onset hereditary spastic paraplegia: a single-center study in Japan
Hereditary Spastic Paraplegia (HSP) is one of the hallmark diseases associated with KIF1A mutations, and we’ve discussed KAND & Spasticity before. But there are over 80 types of HSP, many of which are associated with mutations in specific genes, so observing spasticity alone isn’t sufficient for a diagnosis. How common are KIF1A mutations among children with spasticity, and what other symptoms might point to KIF1A as a possible culprit?
In this week’s article, researchers in Japan investigated the genetics of hereditary spastic paraplegia in a retrospective study: this means they went back and analyzed existing data, in this case the genetics and clinical profiles of 37 children diagnosed with HSP.
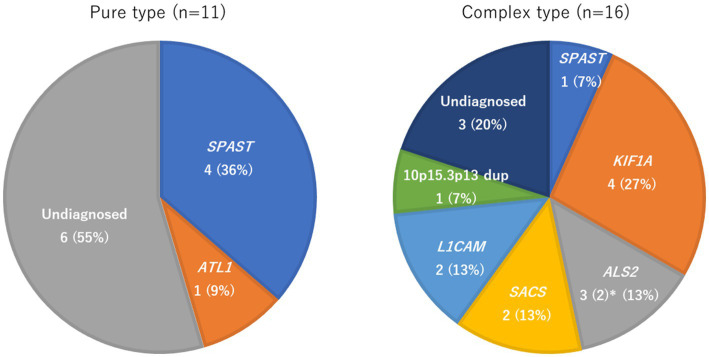
The authors started by breaking down the 37 cases of HSP into two categories:
- Pure HSP describes lower limb spasticity, sometimes accompanied by bladder issues but without other symptoms.
- Complex HSP describes lower limb spasticity with other neurological symptoms, including visual and cognitive deficits.
Only some of the patients had undergone genetic testing: 11 with pure HSP, and 16 with complex HSP. Among the complex cases, KIF1A was the most commonly mutated gene: 4 of the 16 patients carried KIF1A mutations, and all of these mutations were in KIF1A’s motor domain, a region associated with more severe symptoms.
This study has a small sample size from a single hospital, and there were patients who did not have available genetic data, so we can’t generalize these statistics too much. But it does support looking for KIF1A mutations in patients with undiagnosed HSP. The study also noted that three of the four patients with KIF1A mutations also had cerebellar atrophy identified by MRI – Complex HSP with cerebellar atrophy may be a good indicator to test for KIF1A mutations.
Our community has grown quite a bit in the last few years, but we often talk about how KAND is still likely under-diagnosed due to its overlap with other disorders. Integrating KIF1A genetic testing into the diagnostic process for these disorders will help to identify KAND in the broader population.
Rare Roundup
Cautious Optimism for Newly Approved Drug for Rare Form of Genetic ALS
When is there enough evidence for a drug that we can call it a treatment for a disease? This question permeates every step of the drug development process, from testing in cells and animal models to clinical trials and regulatory approval. It’s important to show that a drug works as intended to improve the lives of patients, and does so with reasonable safety.
In past Roundups we’ve discussed how the United States FDA is implementing new strategies to accelerate therapeutic development for rare diseases, reflecting a shift in the culture of drug regulation. Those strategies are now being put into practice for the amyotrophic lateral sclerosis (ALS) community.
Like in our spasticity discussion above, ALS can be caused by a number of gene mutations, with SOD1 mutations responsible for an estimated 2% of cases, called SOD1-linked ALS. Tofersen, an antisense oligonucleotide (ASO), is intended to knock down the toxic mutant copy of SOD1, allowing the healthy copy to function. After its Phase 3 trial, tofersen has been approved as a treatment for SOD1-linked ALS.
This is in part due to the implementation of the FDA’s Accelerated Approval program. In many cases, drug approval relies on a clinical trial’s primary endpoint. This is a single clinical measurement, chosen before the trial begins, that the researchers hope to impact with drug intervention. This constraint can be difficult, especially in multisystem disorders that have a wide variety of symptoms or heterogeneity between patients.
In its recent trial, tofersen treatment did not appear to significantly impact its primary endpoint, the patients’ score on the ALS Functional Rating Scale. However, it did show a number of other benefits in the patient population, including something the FDA hasn’t traditionally considered in its approval process: Biomarkers.
Biomarkers are measurements of biological differences between disease patients and the healthy population, like the presence or absence of certain proteins in the blood. Biomarkers are often a byproduct of the underlying disease, but might not contribute to the symptoms the patient experiences. For this reason they are called a surrogate, or indirect, endpoint.
In SOD1-linked ALS patients, two biomarkers responded to tofersen treatment:
- SOD1 levels in the spinal fluid of patients dropped in response to tofersen, indicating that it knocks down mutant SOD1 as intended.
- Neurofilament light chain (NFL) is a part of the skeleton of neurons, and when it is found in blood it usually indicates the death of neurons. NFL levels were higher in SOD1-linked ALS patients and dropped in response to tofersen, indicating a potential impact on neurodegeneration.
In consideration of these biomarkers and other secondary endpoints, the FDA approved tofersen even though it failed its primary endpoint, accelerating the use of tofersen by patients with SOD1-linked ALS. This holistic approach could be applied to many other rare disorders, but requires continued evaluation of these treatments as they reach the market and patients in need.