

How could small molecule drugs help treat KIF1A-Associated Neurological Disorder? This blog post and accompanying video was created by a team of biotechnology students at Columbia University as part of a class project for the Seminar in Biotechnology taught by Dr. Lili Yamasaki. Thank you to Yanzhe Ma, Yishu Yin, Jerry Shen, Alex Jin, and Wanyun Li for volunteering your time and expertise to create this Research Simplified resource for the KIF1A.ORG community!
In a previous blog post, we introduced what small molecules drugs are. A common follow-up question is what small molecule drugs can do to treat KIF1A-Associated Neurological Disorder (KAND). In this blog, we are going to talk about the potential of small molecule drugs for KAND.
To understand the implication of small molecule drugs for KAND, we have to start with the general mechanism of KAND, a genetic disorder—meaning the disease is caused by changes in the patients’ DNA, which is very important genetic material. A gene is a segment of the DNA that codes for something. We can think of a gene as a blueprint that instructs our body to build things based on its instructions. Without the right blueprints, our body cannot make correct products. Without correct products, the human body cannot function properly. In the case of KAND, the blueprint for an important protein KIF1A (KIF1A gene) changed a little. As a result, KIF1A proteins are built incorrectly and lead to KAND. Normally, KIF1A proteins are responsible for transporting different important cargos for the health of neurons [1]. Imagine trucks traveling on highways to deliver cargos; KIF1A proteins are like those trucks. These proteins can pick up the desired substances and move on structures called microtubules to make deliveries inside cells. But when there are some changes in the DNA of KIF1A protein, our body can no longer produce a functionally correct protein. To use our truck analogy, the truck might drive too slow, too fast, or not make it on the road at all—or have other problems that prevent it from delivering its cargo.
These issues can cause KAND. Clinically, the commonly associated symptoms of KAND include hereditary spastic paraplegia, optic nerve atrophy, epilepsy, ataxia, hypotonia, intellectual disability, cerebellar atrophy, peripheral neuropathy, autism, and attention deficit disorder [2]. The different changes in the KIF1A gene alter the functions of the KIF1A protein in different ways. As a result, an individual with the disorder does not necessarily exhibit the same symptoms as other patients. This is partly why the disease progression varies from person to person, even with those sharing the same KIF1A variant. As we mentioned above, KAND is a genetic disease. To develop a drug against a disease, scientists may target different steps in disease development. In KAND treatment, one can think of two approaches: treating the various symptoms or fixing the KIF1A protein. Still, at the current stage, small molecules are limited to symptomatic treatment options.
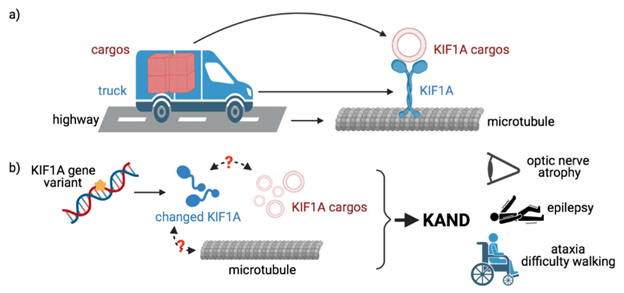
(a) KIF1A functions as a truck to transport cargos
(b) KIF1A variants lead to impaired transportation function and cause KAND symptoms
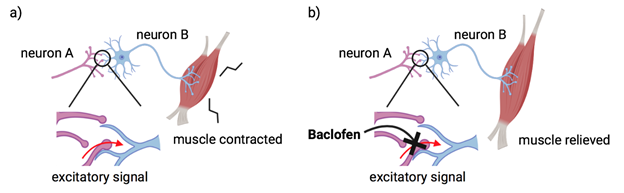
Though current small molecule drugs may not cure KAND, small molecule drugs have large implications in treating KAND symptoms. Taking the drug Baclofen as an example, some KAND patients commonly take this drug to treat spasticity, which results from hyperactive muscles. Though the drug cannot cure the condition, Baclofen can help relax the body’s muscles and relieve the pain [3]. Once the Baclofen enters the body, it binds to the GABA β receptor to inhibit certain signaling and relieves muscles [4]. Keppra, also known as Levetiracetam, is another small molecule drug taken by some KAND patients to treat epilepsy [5]. The drug binds to the SV2A protein that protects against seizures [6].
In the above paragraphs, we emphasized that, currently, there are no small molecule drugs to cure KAND. However, small molecule drugs are a very robust field with much ongoing research. Currently, there are clinically available small molecule drugs to treat other genetic diseases such as spinal muscular atrophy (SMA) and cystic fibrosis (CF). By examining the mechanisms of these new small molecule drugs, we can get a better sense of the development in the small molecule drugs and the insights that may be adapted into therapies for KAND.
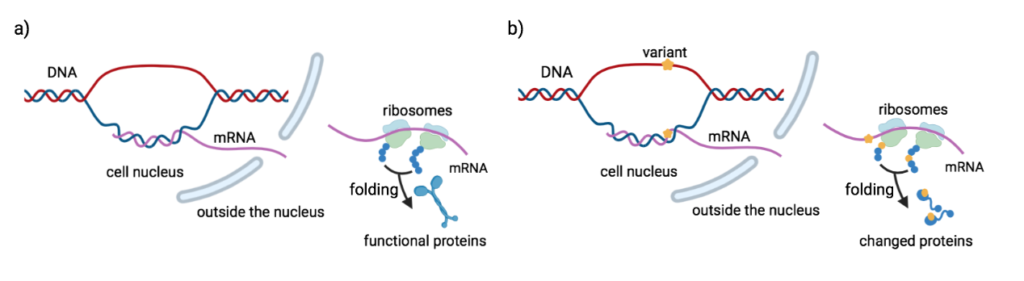
We previously talked about DNA containing important genetic information and how our body can use these as blueprints to build proteins. But how exactly are these proteins built? It all has to begin with DNA. Every cell has DNA. DNA is located at the nucleus of the cell and never leaves the nucleus. However, the ‘machine’ used to make protein, the ribosome is located outside of the nucleus and is unable to enter the nucleus. Thus, cells need messengers to travel from the nucleus to the ribosome and pass the instructions to make proteins. These messengers are called messenger RNA (mRNA). mRNA carries the instructions from the DNA and tells the ribosome what to make. In biology, we call this process of making protein, protein synthesis.
A genetic disorder is caused by changes in one’s DNA. mRNA then carries the incorrect instructions from the DNA to the ribosome. Since the ribosome receives the wrong instructions, it makes an incorrect version of the protein. To treat genetic disorders, scientists can correct the DNA changes so that the mRNA can carry the correct instructions to the ribosome to make functionally correct protein. Another way to treat genetic disorders is to alter the mRNA. In this example, the altered mRNA carries the correct instructions to the ribosome. Though the DNA is incorrect, ribosomes can make the correct protein based on the instructions given by the altered mRNA. Scientists can also target functionally incorrect protein directly, without changing the DNA and mRNA.
Evrysdi is the small molecule drug used to treat SMA, a serious and progressively fatal neuromuscular disorder [7, 9]. The disease is caused by changes in the SMN1 gene [7, 8, 9]. Consequently, the SMN1 gene is unable to produce sufficient SMN proteins [7, 9]. Lack of sufficient SMN protein leads to the disease [7, 9]. Interestingly, scientists have found another gene in our body called SMN2 [7, 9]. Naturally, this gene can produce SMN protein at a low level [8]. This low production level of the protein is caused by certain codes in the SMN2 gene [7, 9]. Instead of targeting the SMN2 gene directly, Evrysdi alters the mRNA to produce sufficient proteins [7, 10]. What does this mean for patients? In clinical trials for Type 1 SMA, some infants were able to sit without support—which is not typical of SMA patients—and the drug helped increase the survival rates for a majority of patients [11]. Worth noting, Evrysdi CANNOT be used to treat KAND. However, the drug’s mechanism provides insight into how a small molecule drug might treat KAND at the genetic level.
Evrysdi provides an example of how small molecule drugs may function to target mRNAs. Some other small molecule drugs can directly target the dysfunctional proteins. Small molecule correctors are one of the examples. With the binding of small molecule correctors, the misfolded proteins that arose from incorrect codes of genes may be arranged into a correct structure and restore protein functions. A majority of patients with cystic fibrosis have misfolded proteins due to the changes in the gene. And those misfolded proteins can be fixed with these small molecule correctors [12]. CF drugs containing small molecule correctors like Orkambi and Symdeko have already been approved for clinical use [12,13]. Both of the drugs have been proved useful to prevent lung exacerbation. Take Symdeko as an example, CF patients taking the drug experienced a 4% increase in lung function after 24 weeks of treatment [14]. The result is significantly effective compared to some other treatments. It is hypothesized that some KAND patients also have misfolded proteins [15]. Hence, CF correctors may provide some insights into KAND drug development for these KAND patients.
How do we identify potential small molecule drugs for KAND? One approach is to use technology to screen thousands of existing small molecule drugs that might be repurposed to treat KAND. This is the objective of a KIF1A.ORG funded high-throughput drug screening with the Christodoulou Lab at Murdoch Children’s Research Institute in Australia. You can learn more about this project here. Without a doubt, advancements in KAND research and small molecule drug development steadily pave the way for a promising small molecule KAND drug to come into the market.
Questions? Leave a comment or send an email to impact@kif1a.org!
All figures were created with BioRender.com
References:
- Boyle, L., Rao, L., Kaur, S., Fan, X., Mebane, C., Hamm, L., . . . Chung, W. K. (2021). Genotype and defects in microtubule-based motility correlate with clinical severity in KIF1A-associated neurological disorder. Human Genetics and Genomics Advances, 2(2), 100026. doi:10.1016/j.xhgg.2021.100026
- Signs & symptoms. (2021, January 14). Retrieved March 21, 2021, from https://www.kif1a.org/kif1a-gene/signs-symptoms/
- Baclofen (oral Route) description and brand names. (2021, February 01). Retrieved March 24, 2021, from https://www.mayoclinic.org/drugs-supplements/baclofen-oral-route/description/drg-20067995
- Bowery, N. (2016). Baclofen: Therapeutic Use and Potential of the Prototypic GABAB Receptor Agonist.
- Levetiracetam (oral Route) before using. (2021, February 01). Retrieved March 24, 2021, from https://www.mayoclinic.org/drugs-supplements/levetiracetam-oral-route/before-using/drg-20068010
- Ciruelas, K., Marcotulli, D., Sullivan, J. M., & Bajjalieh, S. M. (2019). Levetiracetam inhibits sv2a-synaptotagmin interaction at synapses that LACK SV2B. doi:10.1101/640185
- Sheridan, C.. (2021). First small-molecule drug targeting RNA gains momentum. Nature Biotechnology, 39(1), 6–8. http://doi.org/10.1038/s41587-020-00788-1
- Stevens, D., Claborn, M. K., Gildon, B. L., Kessler, T. L., & Walker, C.. (2020). Onasemnogene Abeparvovec-xioi: Gene Therapy for Spinal Muscular Atrophy. Annals of Pharmacotherapy, 54(10), 1001–1009. http://doi.org/10.1177/1060028020914274
- Kolb, S. J., & Kissel, J. T.. (2015). Spinal Muscular Atrophy. Neurologic Clinics, 33(4), 831–846. http://doi.org/10.1016/j.ncl.2015.07.004
- Campagne, S., Boigner, S., Rüdisser, S., Moursy, A., Gillioz, L., Knörlein, A., … Allain, F. H.-T.. (2019). Structural basis of a small molecule targeting RNA for a specific splicing correction. Nature Chemical Biology, 15(12), 1191–1198. http://doi.org/10.1038/s41589-019-0384-5
- Evrysdi helped infants achieve a key motor milestone and survive without permanent breathing support. (n.d.). Retrieved April 03, 2021, from https://www.evrysdi.com/about-evrysdi/type-1-sma-study-results.html
- Lopes-Pacheco M. (2020). CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Frontiers in pharmacology, 10, 1662. https://doi.org/10.3389/fphar.2019.01662
- CFTR Modulator Types. (n.d.) Retrieved March 24, 2021 from https://www.cff.org/Research/Developing-New-Treatments/CFTR-Modulator-Types
- SYMDEKO® RESULTS. (n.d.) Retrieved March 24, 2021 from https://www.symdeko.com/clinical-studies
- Kaur S, Van Bergen NJ, Verhey KJ, Nowell CJ, Budaitis B, Yue Y, Ellaway C, Brunetti-Pierri N, Cappuccio G, Bruno I, Boyle L, Nigro V, Torella A, Roscioli T, Cowley MJ, Massey S, Sonawane R, Burton MD, Schonewolf-Greulich B, Tümer Z, Chung WK, Gold WA, Christodoulou J. Expansion of the phenotypic spectrum of de novo missense variants in kinesin family member 1A (KIF1A). Hum Mutat. 2020 Jul 11:10.1002/humu.24079. Doi: 10.1002/humu.24079. Epub ahead of print. PMID: 32652677; PMCID: PMC7908811.
Fantastic presentation of the latest information. Thank you!
Please keep working on this. Kif1A families are relying on all the hardworking scientists.
We are so hopeful that a breakthrough is just on the horizon.