

#ScienceSaturday posts share exciting scientific developments and educational resources with the KAND community. Each week, Dr. Dominique Lessard and Dr. Dylan Verden of KIF1A.ORG summarize newly published KIF1A-related research. From February 5 – April 16, 2022, a team of talented students from Columbia University’s M.A. in Biotechnology program is taking over the Rare Roundup section of the #ScienceSaturday blog! What topics do you want to learn more about? Send suggestions to our team at impact@kif1a.org.
KIF1A-Related Research
A neuropathy-associated kinesin KIF1A mutation hyper-stabilizes the motor-neck interaction during the ATPase cycle
This week’s #ScienceSaturday paper highlights an E239K KIF1A variant that has been identified in a family with axonal-type Charcot-Marie-Tooth disease as well as in 24 other cases of human neuropathies. This study focuses on the story of two brothers between 60 and 70 years of age who have both experienced late onset, yet progressive, neurological symptoms such as muscle atrophy, weakness, and balance difficulties. When a genetic cause of these symptoms was investigated, it was revealed that both brothers had a hereditary (passed down from a parent) E239K KIF1A mutation. After revealing the clinical presentation of this KIF1A variant, the authors further investigated how and why this E239K mutation could impact the KIF1A protein.
From this paper, we learn that the E239K mutation significantly impairs the transport of common KIF1A cargos in axons by causing the mutated KIF1A protein to move slowly on the microtubule tracks. How is this E239K mutation causing KIF1A proteins to slow down their cargo transport? It turns out that while KIF1A can still engage with the microtubule tracks, the way in which KIF1A utilizes cellular fuel is not optimal. In other words, this E239K mutation is affecting the “engine” of the KIF1A motor, causing it to not use “gasoline” as efficiently as a non-mutated KIF1A motor. Additional work in this paper goes on to describe why this is happening at a structural level, increasing our understanding of how small changes in KIF1A protein shape can lead to large scale clinical outcomes. Want to learn more kinesin basics? Check out this video!
Rare Roundup
Welcome to the #ScienceSaturday Takeover portion of today’s post! Meet our guest bloggers from Columbia University, Aaron, Pragya, Keyue, Rakshitha, and Hazel, here.
Discovering Biomarkers for Neurological Disorders
As we march toward the first clinical trials for KAND, biomarkers are becoming even more important to the KIF1A.ORG family and scientific community. So what are they? In this video below, Dr. Janet Woodcock of the U.S. FDA states simply: “biomarkers are characteristics of the body that you can measure.” Discovering new biomarkers is a crucial step in both diagnosing disorders as well as evaluating potential treatments, particularly for rare diseases like KAND. Biomarkers are used in clinical trials to measure how well the body responds to the potential treatment. Examples of biomarkers include your basic blood pressure and heart rate, but also more complex markers within our bodies such as the proteins neurofilament light chain (NfL) and tau. Dr. Woodcock stresses how crucial it is to improve the clinical trial process and success rate by identifying novel biomarkers so scientists can tell earlier on whether an investigational drug is safe and effective.
Let’s have a look at how researchers can use biomarkers in epilepsy and neurodegenerative disorders.
Advances in the Potential Biomarkers for Epilepsy: First, a review by Kobylarek et al. highlights potential biomarkers that may be used in neurodegenerative disorders, specifically for patients with epilepsy. This review specifies various biological factors contributing to epilepsy, and suggests that hypoxia and oxidative stress may both be causative factors for the onset of epilepsy. HMGB1, a factor that leads to oxidative stress, increases post-epilepsy, and could be used as a potential non-invasive biomarker to identify patients with a high risk of epilepsy. The authors also describe studies based on microRNA, protein and amino acid profiles, which can be detected from biofluids. Their summary of proteins leading to the onset of epilepsy, together with their corresponding therapeutic strategies, is a great way to comprehend the various ways in which epilepsy could be triggered and treated, which may differ for each individual. This may be an excellent opportunity for diagnosis and therapeutics, since the analysis of biofluids is minimally invasive to patients.
Large-scale MS biomarker study: The Cleveland Clinic Neurological Institute launched a new study to uncover early-stage biomarkers for neurological diseases, which will help identify and prevent illness before the asymptomatic stage. Researchers plan to build up a database of 200,000 individuals who are so far neurologically healthy and track their data over 20 years. It is usually too late for us to investigate the initial development of neurological disorders when the symptoms are apparent. However, proactive data collection of biomarkers on such a vast scale will give us new tools to understand disease progression early. The Cleveland Clinic Brain Study will enroll 10,000 volunteers for the first phase, looking for neurologically healthy adults above the age of 50, and neurologically healthy adults above the age of 20, who have a close relative diagnosed with multiple sclerosis. They will go through extensive assessments, and advanced computer tools will analyze their data to find the genetic risk factors and map “disease fingerprints”—biomarkers that can be used for early detection and treatment.
Epilepsy Genetics and Precision Medicine in Adults: A New Landscape for Developmental and Epileptic Encephalopathies
Many in our community understand the frustrating journey to a correct diagnosis of KAND. Even after diagnosis, patients may be surprised to find out that there is no single symptomatic landscape for KAND—every person is different, and so are the particulars of their condition. While KAND is defined by mutations in the KIF1A gene, the location and nature of the mutation can vary widely, and this can also impact potential gene-based therapies. A recent review by Beltrán-Corbellini et al. discusses how different genetic mutations can manifest in epileptic encephalopathies, and how they impact approaches to diagnostics and treatment.
While the paper does not discuss KAND specifically, it does very clearly explain genetic and scientific concepts that impact our community.
Types of Mutations
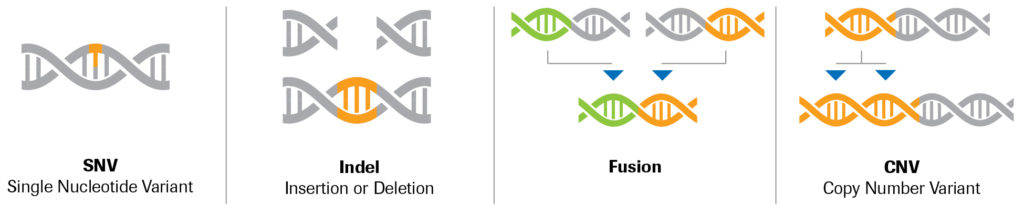
KAND can be caused by a variety of mutation types, including:
- Single Nucleotide Variants (SNVs): A single nucleotide in a DNA sequence is substituted with a different type of nucleotide. These mutations may result in slight alterations in the KIF1A protein that can have various impacts on its function, some subtle and some severe. They are the most common mutations in KAND.
- Insertions/Deletions (Indels): Small (1-49 nucleotide long) insertions or deletions in a DNA sequence. While these may have a similar effect to SNVs, indels can also have more profound consequences—amino acids, the building blocks of proteins, are made by triplets of nucleotides, and an indel that disrupts this pattern can alter everything downstream of the mutation.
- Structural Variants: Insertion, deletion, duplication, translocation or inversion of segments of DNA more than 50 nucleotide in length, sometimes reaching chromosomal scales. Such mutations may impact not only the KIF1A gene, but other nearby genes as well.
The paper also discusses complexities of genetic disorders, such as:
- Phenotypical heterogeneity: two patients affected by the exact same mutation can have different symptoms
- Genotype-phenotype correlation: while researchers try to predict the set of symptoms a patient experiences (phenotype) based on their mutation (genotype), this correlation can be difficult to make, especially in progressive genetic disorders that change over time
- Mosaicism: While rare, unaffected parents may carry a mutation in a small percentage of their cells and pass the mutation to their child(ren), who develops the genetic disorder—this article highlights the need for genetic testing that is capable of identifying these mosaic cases
Rare Diseases: Maintaining Momentum
A new report from Global Genes (a leading rare disease advocacy organization, of which KIF1A.ORG is a member) stated that in 2021, US$22.9 billion was invested in drug development on rare disorders, a 28% increase from 2020. Similarly, research progress has been gaining momentum. For example, the International Rare Disease Research Consortium (IRDiRC) aimed to achieve two main objectives by 2020: to diagnose most rare diseases and to deliver 200 new therapies. Diagnosing most rare diseases is within reach, and the development of 200 new therapies was achieved in 2017! Looking to the future, the IRDiRC have set new goals for the coming decade: for 1000 new therapies for rare diseases to be approved, and for all patients with a suspected rare disease to be diagnosed within a year, or enter a research study to determine diagnosis.
In achieving such goals, collaboration is crucial. The European Reference Networks have been vital in sharing knowledge between healthcare professionals across borders, improving the standard of care. Nonetheless, The Lancet calls for a new policy framework that would build upon existing European health and research programmes to guide the implementation of national strategies across all countries in Europe. EURORDIS-Rare Disease Europe, a non-profit alliance of 962 rare disease patient organizations from 73 countries, is campaigning for such a trans-European action plan. Thus, there is significant momentum currently in the rare disease space. However, action is needed to make sure this is maintained in Europe, with The Lancet underlining the importance of trans-European cooperation.