

#ScienceSaturday posts share exciting scientific developments and educational resources with the KAND community. Each week, Dr. Dominique Lessard and Dr. Dylan Verden of KIF1A.ORG summarize newly published KIF1A-related research and highlight progress in rare disease research and therapeutic development.
KIF1A-Related Research
Genetic and clinical landscape of childhood cerebellar hypoplasia and atrophy
When we discuss KAND treatments, be they small molecule or RNA/DNA-based therapies, one of our biggest questions is: How will this impact “X” symptom? In most cases, the unfortunate answer is that we don’t know. One major factor in this uncertainty is that KAND is both a neurodevelopmental disorder, and a neurodegenerative disorder. This means that if we see that a brain region isn’t formed or behaving correctly, there are three possibilities:
- Neurodevelopmental: The brain region didn’t form normally during development, a symptom called hypoplasia (hypo=less, plasia=growth).
- Neurodegenerative: The brain region formed normally and then began to degenerate, a symptom called atrophy.
- Combination: The brain region didn’t form normally, and then degenerated as well.
While these causes may result in similar symptoms, they may respond to different treatments or have different therapeutic windows: Neurodevelopmental symptoms may require a much earlier intervention than neurodegenerative symptoms, for example.
Among our families, a common symptom is cerebellar pathology. The cerebellum (or “little brain”) sits near the base of our skull and is primarily responsible for fine-tuning our movement, including walking. But because brain scans aren’t regularly performed on asymptomatic children, it can be difficult to tell whether the cerebellum developed abnormally, began to degenerate, or both. In this week’s study, researchers investigated 188 patients with cerebellar pathology to better distinguish hypoplasia from atrophy.
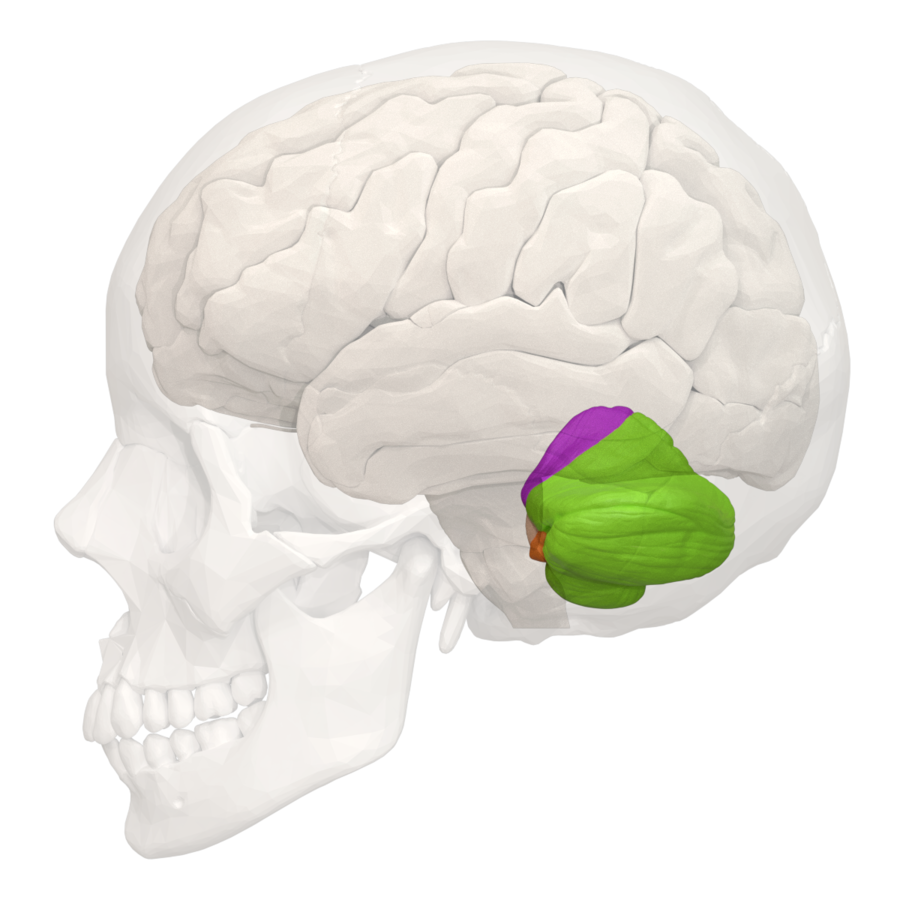
Because different genes can contribute to similar symptoms, the authors performed genetic sequencing to look for mutations. Disease causing variants were identified in 105/188 patients, including 8 patients with KIF1A mutations. Notably, the degree of cerebellar loss was highly variable within the KIF1A group, likely reflecting mutation heterogeneity in our community.
When the authors analyzed genetic variants in their patients, they found that many of these genes are associated with neurodegenerative atrophy. However these genes were also specifically upregulated during childhood and adolescent development. This finding suggests a potential overlap between neurodevelopment and neurodegeneration. Further clinical assessments will be needed to distinguish the two, especially in variable patient populations like KAND.
Distinguishing between similar types of brain dysfunction is incredibly complex, and this study represents an early step to integrating genetic data into diagnosis. But in the long-term, dissecting these symptoms is important. As the authors state: “If clinicians know that aberrant genes are atrophy-related in patients… their clinical management may become more cautious.” We look forward to following this work in the future to learn more about KAND.
Rare Roundup
UK Government launches Newborn Genomes Programme
One of the biggest challenges faced by rare disease patients is the prolonged diagnostic journey, when families watch symptoms develop without knowing the cause. This undiagnosed period is a bottleneck that limits symptom management and treatment, and leaves families in a state of uncertainty.
An important factor in diagnosis is genetic testing: Undiagnosed patients often undergo genetic panels to look for mutations associated with their symptoms. But these panels may not include all relevant genes, and often take place after patients have been suffering from symptoms for extended periods of time. But what if we were to get a head start by gene sequencing newborns, including healthy ones? This is the question being asked by the UK National Health Service’s Newborn Genome Programme.
The project will entail whole genome sequencing of up to 200,000 newborns in the UK. The goal is to determine potential health-altering variants before children experience disease, so that parents and clinicians are better prepared to take action quickly if symptoms arise.
By performing this screening at such a large scale, the study aims to address several unanswered questions regarding the use of whole genome sequencing in disease diagnostics and healthcare, as described by Genomics England:
- How will it integrate with the NHS newborn screening blood spot test?
- How conclusively can it predict disease in pre-symptomatic babies, and how does it impact their diagnostic experience?
- How should it be paired with other screening and diagnostic tools?
- How to best guide parents through that uncertainty to avoid unnecessary ‘medicalisation’?
The study also acknowledges that genetic information can be ambiguous and overwhelming for patient families, and seeks to integrate holistic information about patient experience to provide better guidelines for genetic diagnostics. This represents a huge step toward more comprehensive and informed screening guidelines to identify, manage, and treat rare diseases as early as possible.