

#ScienceSaturday posts share exciting scientific developments and educational resources with the KAND community. Each week, Dr. Dominique Lessard and Dr. Dylan Verden of KIF1A.ORG summarize newly published KIF1A-related research. From February 5 – April 16, 2022, a team of talented students from Columbia University’s M.A. in Biotechnology program is taking over the Rare Roundup section!
KIF1A in the News
The human genome has finally been decoded. Here’s why this discovery is a game changer: An interview with Dr. Wendy Chung
This week Dr. Wendy Chung, a pillar of the KIF1A research community, discussed strides in gene sequencing technology and her hopes for gene editing technology to treat rare diseases in a CNN+ interview. The full interview (available only to CNN+ subscribers) included KIF1A superhero Susannah and her parents, Luke Rosen and Sally Jackson, co-founders of KIF1A.ORG. We thank Dr. Chung and Team Susannah for their relentless efforts to raise awareness and drive research forward!

KIF1A-Related Research
ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function
This week we return to a topic we explored in our March 12th #ScienceSaturday: KIF5A gain-of-function mutations.
KIF5A is another kinesin motor protein whose structure is similar to KIF1A: it forms in pairs to carry cargo along neuronal axons, and mutations can result in amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disorder.
As we discussed last month, KIF5A proteins usually stay dormant until binding to another KIF5A as well as a cargo, a process called autoinhibition. Mutations can alter the part of KIF5A responsible for this autoinhibition, making the protein active at all times, which can cause KIF5A to accumulate away from the cell body. Imagine running a distribution center where your semi-trucks left whether they had cargo or not—pretty soon your warehouse would be empty and unable to make shipments!
This week’s featured article further assessed KIF5A gain-of-function mutations using a variety of cell lines, including patient-derived neuronal cultures. The study found that mitochondria, which provide energy for cellular processes, move faster along axons when carried by mutant KIF5A, which could underlie energetic failure and neurodegeneration seen in ALS.
Papers like these highlight an important fact about kinesin mutations: where the mutation occurs can have a big impact on function! While alterations to the motor domain of kinesins can cause a decrease in transport, changes to regulatory domains (like those responsible for autoinhibition) can cause increases in transport, and both can have negative consequences for neurons.
Rare Roundup
Welcome to the #ScienceSaturday Takeover portion of today’s post! Meet our guest bloggers from Columbia University, Aaron, Pragya, Keyue, Rakshitha, and Hazel, here.
Pathophysiology of neurodegenerative diseases: New approaches for investigation and recent advances
De novo mutations in KIF1A are very common. In the latest publication by Boyle et al. of the KAND natural history study (funded in part by KIF1A.ORG!), among 67 patient-parent trios, only 3/67 patients inherited the mutation from their parents, whereas the remaining 64/67 all developed the mutation spontaneously. In a single case, the patient was mosaic for the mutation in blood. Since the paper was published last year, we now know of at least five parents who are mosaic for KIF1A, but mosaic cases are still rare. De novo mutations are also more likely to cause severe types of KAND.
This paper discussed the detection, causes and impacts of de novo mutations. Our cells keep dividing throughout development and sometimes they make mistakes when making DNA copies, spontaneously leaving a mutation in our genome in a subset of our cells. This mutation is called a de novo (“from new”) mutation, as it is made from scratch and not inherited from our parents. Only mutations present in the germline (cells giving rise to sperm or eggs) can be passed down to the next generation. About 89% of de novo mutations arise solely in the germline of parents (as shown in the left of the picture). This is because both sperm and egg cells require rounds of cell division to be mature and fertile, during which mutations are likely to occur. 7% of de novo mutations arise after the egg gets fertilized and becomes an embryo (as shown in the right of the picture). Since not all the cells will carry the mutation, a phenomenon called mosaicism, the influence of the mutation will be restricted to specific types of tissues and organs depending on the timing. In some cases, a disease-causing mutation does not affect the individual at all because it shows up in the wrong organs where the gene does not function. However, some unaffected individuals have this mutation in their germline and can transmit it to their kids, leading to the remaining 4% of de novo variants (as shown in the middle of the picture). Unlike mutations in the germ cell alone, this type of mutation is also present in other tissues and detectable in the blood of the parents.
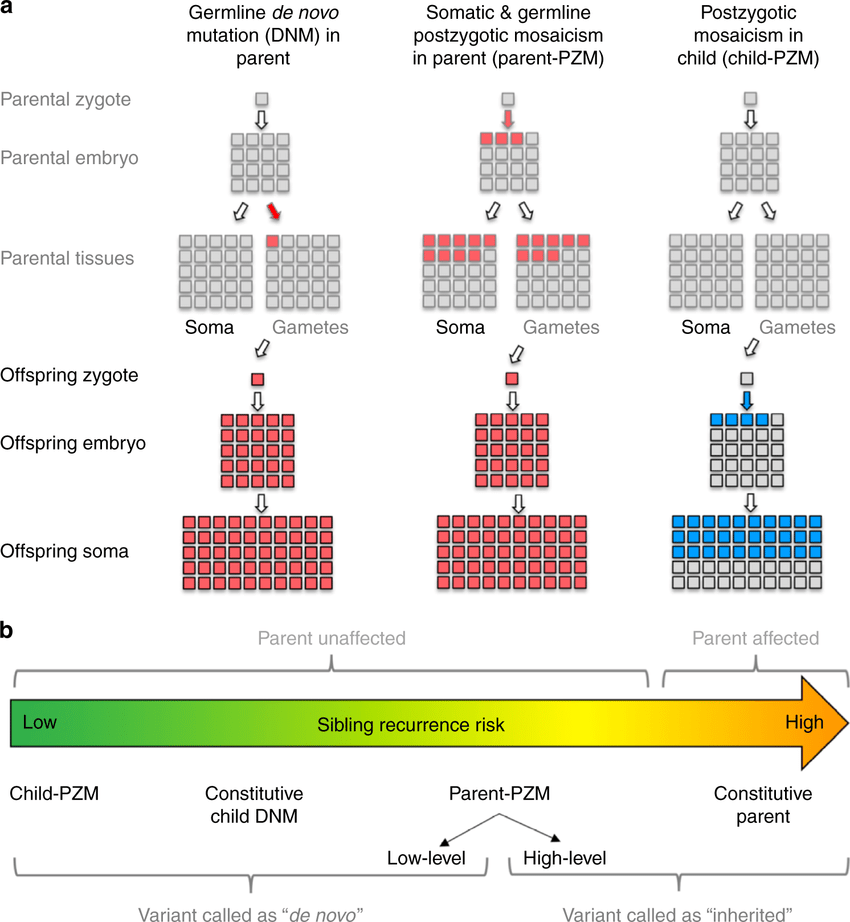
We can determine whether a mutation is inherited or de novo by sequencing both the affected individuals and their parents. To maximize its ability to identify a de novo variant, the sequencing approach should be high in quality and coverage. High quality means that it faithfully reflects your genetic information, instead of making errors by itself that could be mistaken as a mutation. High coverage means many copies of a gene are sequenced, which could increase the possibility of capturing the small fraction of mutations in a mosaic individual.
Improving the targeted treatment of movement disorders: Optimizing deep brain stimulation in patients with dystonia
This article by Charité – Universitätsmedizin Berlin discusses the recent discoveries that may prove vital in improving the nonchemical treatment of Dystonia—a rare neurological disorder characterized by involuntary, twisting, and distorting movements. People with dystonia may be limited in their ability to perform activities such as drinking, walking, and speaking. The condition is subdivided into generalized dystonia, which affects the entire body, and focal dystonia.
Experts assume that symptoms are a result of faulty interactions between areas of the brain that lead to abnormal signal transmission. One treatment option available to patients is a neurosurgical procedure involving the implantation of electrodes into specific areas of the brain. Known as deep brain stimulation, the procedure involves the implantation of a pacemaker-like device and is often the only treatment capable of providing relief of symptoms. “The precision with which this stimulation has to be adapted to the symptoms seen in different types of dystonia was not clear until now,” explains study lead Prof. Dr. Andrea Kühn. After analyzing the electrodes’ precise positions, the researchers were able to generate computer models showing which brain networks were being activated in each of the patients investigated. By mapping data on symptom improvements to their network models, the researchers were then able to determine which of the identified networks were crucial to treatment success.
“Due to the paucity of alternative treatment options beyond deep brain stimulation, our findings make an important contribution to improving treatment for dystonia. In the future, we will be able to more deliberately treat specific types of the disorder,” says Dr. Andreas Horn of the Department of Neurology and Experimental Neurology. Therefore, further research into the heterogeneity of movement disorders is crucial for progress towards personalized treatment options.
Challenges facing biobanking and rare disease care in Africa, with a focus on South Africa
Currently, there are no KIF1A.ORG community members in sub-Saharan Africa, a region of 1.14 billion inhabitants, but it cannot be that there are no KAND patients in a region of this size. Part of this discordance could be because of the language barrier(s) present. Another likely reason is the difficulty of getting a rare disease diagnosis in sub-Saharan Africa. This article from 2021 discusses the benefits and challenges of establishing a rare disease biobank in South Africa. Where possible, it extrapolates these messages to rare diseases and congenital disorders in low-middle income countries (LMIC) as a whole. This rare disease biobank, created by The North-West University’s Center for Human Metabolomics (CHM), will be the first rare disease biobank in Africa. Biobanks are a crucial part of establishing infrastructure that enables more research, better diagnosis, and altogether better standard of care for rare diseases.
Creating this crucial tool is accompanied by a number of challenges and considerations. First is that rare diseases are often not prioritized in South Africa. Priority by health care institutions is often given to common communicable diseases such as HIV/AIDS or malaria. The proportion of child deaths due to congenital disorders and rare disease continues to increase, implying that rare diseases and congenital diseases do not get the same attention as other causes of childhood mortality. However, South Africa is undergoing an “epidemiological transition” with focus shifting from the HIV/AIDS pandemic to other diseases (such as congenital ones). As a result, there is a lack of expertise in the rare and congenital disease space. This problem, associated with an epidemiological transition, is shared across many LMICs. Clinical efforts in the rare disease space, such as creating this biobank, can be especially challenging due to lack of funding and prioritization.
Another example of a challenge that faced in establishing this biobank, and establishing better care and diagnosis for rare diseases in general, is the lack of resources and infrastructure in South Africa (as well as other LMICs). For example, unreliable postal services and the requirement for samples to be kept cold can make samples unviable. This hampers diagnosis and collection of samples in more remote areas.
Thus, while there are significant challenges, a rare disease biobank offers the opportunity of breaking the cycle of poor rare disease related services. This article discusses many more factors to consider in the establishment of the continent’s first biobank including the ethical considerations, the laws concerning consent, the socioeconomic factors, and the benefits that it will bring.
Astrocytes and microglia and myelin, oh my: What are glial cells?
When we discuss the central nervous system, we tend to think about neurons: cells with long projections that transmit electrical activity across networks. But these networks aren’t just comprised of neurons. Glial (“glue”) cells describe a variety of cells that play crucial roles in supporting neuronal function. Learn more in the video below, and then check out some advances in our understanding of glial disease research.
Immature astroglia, not neurons, could be potential epilepsy therapy target
The brain has numerous cell types spread across its various regions, contributing to specific functions. The brain’s hippocampal region consists of the dentate gyrus, responsible for memory and regulating emotion. It is found that this particular part of the brain can generate new neural cells in the adult by a process termed neurogenesis. This production helps modify the existing neural circuitry.
So far, medical and surgical strategies for some epilepsy patients are such that it requires the removal of part of the hippocampus to treat them. In the study conducted by the USC research lab, it was found that there could be a potentially non-invasive alternate method to treat this issue. They studied the living brain tissue donated by patients with epilepsy and compared it to a post mortem brain with no known neurological disorders. While both showed potential for neurogenesis, the epileptic brain consisted of another cell type, known as astrocytes, that were not proliferating in the healthy brain. This finding helped them distinguish between the two brains and work on developing a way to use the astrocytes as a target in treating epilepsy.
The immature astroglia in patients with Mesial Temporal Lobe Epilepsy was studied, and it was noted that while there was a sharp decline in neurogenesis over time, astrogenesis remained persistent. The neuronal death was confined to other cell types since the immature astroglia were consistent in their proliferation. This was fascinating to the research team, as the astroglia were previously known only as environmental cells that provided support to adult neurogenesis. According to scientists, its constant presence in patients with long-term epilepsy may be indicative of it contributing to the initiation and modulation of chronic seizures. This new finding has therapeutic potential that has not yet been explored. It could offer an incredible alternative to treating patients with epilepsy by creating new drugs that target the astroglia and combine existing medical strategies to provide a non-invasive treatment.
Replacing Microglia Treats Neurodegenerative Disease in Mice
Microglia play a role in regulating the pathogenesis of many neurological diseases, including neurodegeneration, autoimmune diseases, vascular brain injury, inflammatory diseases, and brain tumors. Scientists view microglia as a potential cell therapeutic target for neurodegenerative diseases such as Alzheimer’s.
Yohei Shibuya et al. recently published a study on microglial replacement assays in the mouse brain. They used chemical drugs to kill existing microglia in experimental mice and then injected circulation-derived myeloid cells (CDMCs) into the hippocampus of experimental mice. Several weeks later, the researchers observed that bone marrow cells had migrated further from the injection site, were evenly distributed throughout the brain, and exhibited a microglia-like differentiated morphology that remain stable after six months. The experiment has shown that it improves the locomotor activity of experimental mice and extends the lifespan of experimental mice.
The researchers suggest that this experiment validates the feasibility of highly effective microglial replacement with CDMCs for cell-based regenerative therapies to restore brain function. Part of the reason this approach works is because microglia are the “surveillance cells” of the central nervous system—they roam around looking for signs of injury or pathology for them to fix, so they’re mobile enough to repopulate the brain. Still, developing a safer microglial replacement strategy is required to reduce the toxicity of chemical drugs.